
Alzheimer’s Disease
Neuroscientist Mark Mattson on tau proteins, free radicals, and the importance of exercise in maintaining cognitive functions
faq | December 6, 2016







Alzheimer’s Disease (AD) is a type of brain disorder in which patients early on develop a problem with short-term memory (STM). For example, they may have difficulty even remembering in a short conversation that they already told someone something and typically repeat something already told earlier. As the disease progresses, problems with circadian rhythms, long-term memory, as well as aggression, and agitation may develop, and then ultimately, they will not even be able to properly recognize their close family members.

Auguste Deter, the historical patient of Alois Alzheimer (Wikipedia.org)
History of Research
A little over 100 years ago, there was a psychiatrist in Germany. He had a patient who developed symptoms of memory loss. She was about 70 and started having STM problems as well as behavioral anxiety. Then, a pathologist looked at her brain under the microscope and noted what we now know as amyloid plaques: the accumulation of amyloid in the brain. And the name of this psychiatrist was Dr. Alois Alzheimer.
Stages of Alzheimer’s disease
The earliest stage in AD is STM loss which does not affect an individual’s ability to function. Many people, when they get old, have similar memory problems, but they don’t have AD. This makes it difficult to know whether someone is going to develop AD because problems with STM, particularly in the elderly, are very common. And this is called mild cognitive impairment, MCI. What will happen is that a person typically goes to neurologists to do cognitive testing, and they can notice if there is MCI. They continue testing, usually once a year, and there are a variety of tests the patient goes through to evaluate their learning and memory abilities. If the person gets worse and worse over 3-4 years, then it strongly suggests that he or she is developing dementia. With STM problems for a number of years, the patients are still able to function well, but as it progresses, then they might need some help in, say, driving a car because they may forget where they are going or why they are going somewhere.

There is an intermediate stage when the patients are still okay at home, they are able to dress, brush their teeth, and eat, but they need constant care because their STM is already gone. They may also develop other medical problems.
In the very late stages, they might forget who they are, although they are still able to recognize the faces of the people they know. They see your face and smile, even at the very end. So, there is some long-term memory preserved even very late.
The most common reason for death in AD is bacterial pneumonia. Because of the problems in their brain, they develop problems with swallowing food, and they may aspirate or breathe pieces of food into their lungs, causing a bacterial infection of the lungs. It typically takes no more than ten years from early MCI to death. Constant care will usually be for 2-4 years.
What happens in the brain
In the brain, what’s going on is that nerve cells die. But, even before this, the connections between them, which are called synapses, don’t work well since they are degenerating and eventually disappear. If you look at the brain of someone who died with AD under a microscope, you will see that many neurons are gone in the brain regions, such as the hippocampus, for example, that are very important for learning and memory. There will be an accumulation of a protein called amyloid. It’s normally produced in all of us, but in the normal brain of a person who doesn’t have AD, at least certainly young people, the amyloid is removed and doesn’t accumulate. The other abnormality that occurs before nerve cells die is that inside nerve cells, there is another protein called Tau, and it will accumulate and essentially impair the ability of the nerve cells to function properly.
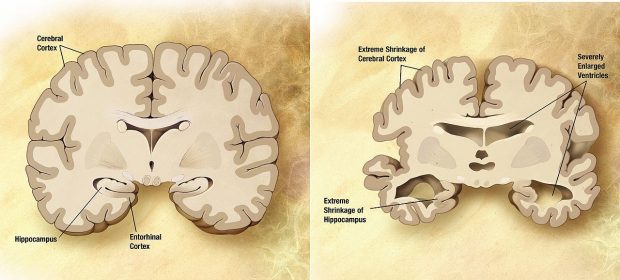
Comparison of a normal-aged brain and the brain of a person with Alzheimer’s (Wikipedia.org)
Amyloid and AD
When someone starts to have STM problems and goes to a doctor to do cognitive testing and brain imaging, they will be diagnosed with probable AD if they meet the criteria. The reason is that about 20-25% of patients who have these symptoms will not have much amyloid in their brain if someone looks in their brain after they die. And since the accumulation of amyloid was early on considered a defining characteristic of AD, then these patients do not have AD. They still have the death of nerve cells and memory problems, but not the amyloid. And we don’t know what is causing the problem, but there are often damaged blood vessels in the brain. And there are people who are 90 years old, for example, and their brain was functioning very well at the time they died, and when we look at the brain under the microscope, they have a lot of amyloids. So, apparently, there are cases of people in whom the nerve cells are resistant to the damaging effects of the amyloid. We are really interested in these cases and trying to understand how their nerve cells are able to tolerate so much amyloid in the brain. In my own lab, we do work with fasting on mice and how mice would accumulate amyloid in their brain. We give them a mutated human gene that causes an early onset inherited AD.
Early-onset familial AD
In a very small percentage of people with AD, a little less than 5%, the disease is caused by genetic mutations. One characteristic of people who inherit the abnormal gene from their parents is the development of symptoms of the disease when they are young, often in their 40s and 50s. There are even families where the patients become affected in their mid or late 30s. It’s terrible because these people often have teenage children, and they have to take care of their own parents.
The symptoms of early AD cannot be distinguished from more common AD, which usually occurs between the ages of 70-80 and 90. We do not know the causes of these most common late-onset cases of AD. However, we do know that many cases of early-onset inherited (familial) AD are caused by mutations in the genes that encode the amyloid precursor protein (APP) and the enzyme presenilin 1 (PS-1), increasing the production of a form of the amyloid that is more prone to aggregation and accumulation in the brain.
Aging and other factors

Lifestyle factors
As far as increasing the risk of AD, the key things that seem to be true are what we in the US call the “couch potato” lifestyles. It is when people do not get any exercise, eat too much, and also don’t keep their minds intellectually challenged. It turns out that if we don’t challenge our bodies and brains with exercise and fasting, or reducing energy and keeping our minds active, then what we think happens is that the nerve cells in the brain become more susceptible to stresses that occur during aging, with a reduced energy level in the neurons.
We have some insight as to why exercise, fasting, and intellectual challenges may protect from AD. All three of these challenges (physical, energetic, or brain exercises) increase the production of proteins that are called nerve cell growth factors. And one of them is called BDNF – brain-derived neurotrophic factor – and tremendous amounts of evidence have shown that it plays a very important role in the brain’s learning and memory skills. BDNF can essentially stimulate the formation of new synapses. Also, these positive lifestyle factors, at least in the hippocampus, can stimulate neurogenesis, which is the production of new nerve cells from stem cells.
Furthermore, it seems that aerobic exercises like distance running, cycling, or swimming are particularly good for the brain compared to weightlifting or yoga, although yoga and meditation can reduce stress. There are some studies on the elderly indicating that their learning and memory can be improved by exercise. In the studies, I know people who were in their 70s, even 80s. They were either put on an aerobic exercise program or just stretching for several months, and their memory was tested before and after these months. So, those who did the exercises had improved learning and memory, whereas the control group that only stretched did not.
But all these factors still cannot prevent anyone from having AD, they only reduce the risk or postpone it.
Treatment
So far there is no treatment that will slow down the progression of the disease. There are many medical trials of drugs that are designed to remove amyloid from the brain or other ones to increase the amount of energy supplied to the neurons or stimulate the production of neural cell growth factors such as BDNF. We are planning to do a study on increasing the energy in the neurons by giving patients a specific ketone. I’ve mentioned we do a lot of work on fasting. Every time you eat a meal, the energy goes into your liver, and it’s stored there. If you eat three meals a day, there is always plenty of energy in your liver, and then the liver releases glucose into the blood, and cells, including the nerve cells in your brain, use glucose for their energy. In AD, the nerve cells have a problem using glucose. When you fast, say, for 24 hours, you completely deplete the energy from your liver, then fats in your fat cells are released into the blood, and they go to the liver where they are converted into ketones, and these ketones are used as an energy source for neurons. In our animal studies, and there is a reason to believe this may be true in humans as well, the ketones can protect nerve cells against dysfunction and even improve cognitive function. So, one approach is to boost the energy levels in the nerve cells. We have some reasons to believe that people who maintain low body weight have a reduced risk of AD. And also, people who have a chronic elevation of glucose levels, i.e. people with diabetes, have an increased risk for AD.
Current research
So, we do research on fasting. In our study, we are taking people who are obese but do not have diabetes between the ages of 55 and 70 and people who are getting old. They are obese and have insulin resistance, which is a precursor to diabetes. We divide them into two diet groups: the first group fasts two days a week, and the other group does not fast. First, we start the diet, and two months later, we test their cognitive functions and look at nerve cell network activity in their brains by MRI. And based on our animal studies, we predict there should be positive changes. Then we are taking cerebrospinal fluid and measuring neurochemicals that we’ve shown to increase in the brain during fasting. We will also be measuring amyloid and Tau in this brain fluid.
Tau protein in neurons (red). (Wikipedia.org)
On the animal study side, we are doing a lot of work with different potential interventions. For example, there is a drug that can have a stimulatory effect on the mitochondria, and in preliminary data, we found that this drug is beneficial. It was used to treat obesity a long time ago, and we think that maybe in the future, this is a potential possibility to strengthen the mitochondria in the nerve cells. Also, in the mice, we found that exercise can increase the number of mitochondria in nerve cells. That’s interesting because it’s very similar to what happens in muscle cells: they get stronger when you exercise, so we found that your nerve cells also get stronger.
Future directions
There are several important areas that we really need to understand better. First, we need to develop some way to determine whether an individual in their 50s is going to have AD because once someone gets symptoms, there are already millions of nerve cells that have died, and that is undoubtedly the main reason why there has been no treatment. By the time the person has symptoms, it’s almost too late.
There is also a lot of interest in the possibility of putting new nerve cells in the brain either by transplanting stem cells in the brain or transplanting immature baby neurons that will grow and form neural circuits. Some labs are trying to figure out if they can cause glial cells in the brain to form neurons. It turns out that there are more glial cells in the brain than nerve cells. Glial cells support the function of nerve cells: they can supply energy, produce growth factors, and so on. And glial cells do not die when a person has AD. It might be possible in the future to stimulate glial cells to form neurons because, unlike nerve cells, glial cells divide. So, several labs have shown that they can make stem cells from a patient’s skin cells and insert two or three genes into the cells that cause them to become neurons. Or convert the glial cells directly into neurons. So far, this is all in a culture dish, but there are technologies that would allow the introduction of these genes that can stimulate glial cells, either directly or indirectly, to form neurons in the brain. This is currently being done in animal models.
So, in the future, it might be possible to replace the nerve cells that have died. And if we do that in the early stages of the disease, we could replace short-term memory cells, and long-term memories won’t be affected. Therefore, the person will be able to form new memories once again. It’s not Sci-Fi; it is theoretically possible based on what we know. It’s just developing the technology and approaches that are necessary to do it safely in a human.




Most viewed













